Screen for X-linked adrenoleukodystrophy (X-ALD) and other peroxisomal disorders from a simple dried blood spot. This LC-MS/MS test measures lysophosphatidylcholine (LPC) derivatives of very long-chain fatty acids — enabling early detection when hematopoietic stem cell transplantation can still halt disease progression.
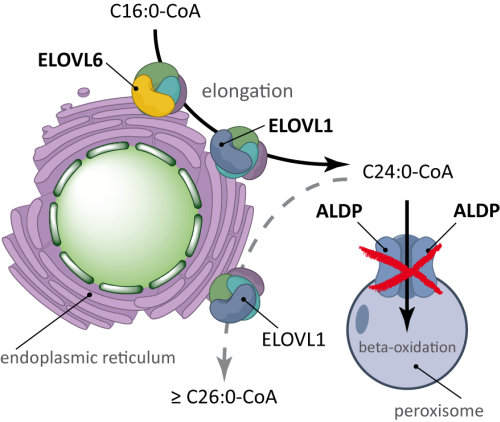
X-linked adrenoleukodystrophy is an inherited metabolic disorder caused by mutations in the ABCD1 gene, which encodes a peroxisomal transporter protein essential for the import of very long-chain fatty acids (VLCFAs) into peroxisomes for beta-oxidation. Without functional ABCD1, VLCFAs accumulate in plasma, red blood cells, and tissues, leading to progressive demyelination of the central nervous system, adrenal cortex dysfunction, and peripheral nerve involvement.
X-ALD affects approximately 1 in 17,000 births. The childhood cerebral form (CCALD) manifests between ages 4 and 10 with rapid, devastating neurological decline. Adrenomyeloneuropathy (AMN) typically appears in the second to third decade with slower progression. Without early detection, the cerebral form causes irreversible loss of motor and cognitive function.
The LPC-VLCFA screening test quantifies lysophosphatidylcholine (LPC) derivatives of very long-chain fatty acids from a dried blood spot using liquid chromatography-tandem mass spectrometry (LC-MS/MS) with stable isotope-labelled internal standards:
LPC-VLCFA measurement in dried blood spots is superior to traditional plasma VLCFA quantification: LPC derivatives are more stable in the DBS matrix at ambient temperature, require minimal sample volume, and have demonstrated higher diagnostic sensitivity and specificity in newborn screening programmes worldwide.
The LPC-VLCFA screening test is designed for B2B partners integrating rare disease screening into their clinical pathways, newborn screening programmes, or diagnostic workflows:
Hematopoietic stem cell transplantation (HSCT) is currently the only accepted therapy capable of halting disease progression in the cerebral form of X-ALD — but only when performed before symptom onset or very early in the disease course, before irreversible neurological damage occurs. This makes presymptomatic detection through newborn screening critically important.
Beyond X-ALD, elevated LPC-VLCFA levels indicate impaired peroxisomal beta-oxidation and can identify a spectrum of related disorders:
Female carriers of ABCD1 mutations may remain asymptomatic or develop a milder neurological syndrome later in life. LPC-VLCFA screening can identify carriers, enabling genetic counselling, family cascade screening, and informed reproductive decisions.
A simple finger-prick or heel prick is used to collect a few drops of blood onto a dried blood spot (DBS) card. The LPC-VLCFA analytes are highly stable in the DBS matrix at room temperature, making this collection method ideal for:
Results are delivered via our secure portal within 3–5 working days of sample receipt and include quantitative LPC-VLCFA values, diagnostic ratios, and interpretive commentary. For branded or white-label reporting, contact us to discuss customisation options for your organisation.
Partner with Fatty Acid Labs for validated LPC-VLCFA screening. Integrate newborn screening for X-ALD and peroxisomal disorders into your clinical pathway with full laboratory support.